Mukopolysaccharidose Typ VII (MPS VII) ist eine autosomal-rezessiv vererbte Stoffwechselkrankheit. Wie bei anderen Mukopolysaccharidosen liegt die Ursache in einem enzymatischen Defizit bei der Verarbeitung von Glykosaminoglykanen (GAGs). Bei der MPS VII fehlt es den Patienten an β-D-Glucuronidase, einem Enzym, das in den Lysosomen für den Abbau der GAGs Dermatansulfat, Heparansulfat und Chondroitin-6-sulfat benötigt wird. Dadurch sammeln sich diese komplexen Kohlenhydrate in verschiedenen Geweben an und können dort zu schweren Schäden führen.
MPS VII wird auch als Morbus Sly bezeichnet, benannt nach Dr. William Sly, der diese Erkankung 1973 erstmals beschrieb. Es handelt sich bei der MPS VII um eine sehr seltene Mukopolysaccharidose.
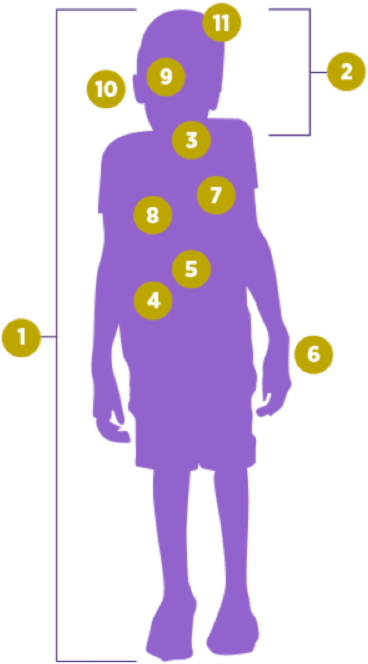
Die Symptome von Patienten mit MPS VII haben eine gewisse Ähnlichkeit mit denen der MPS I. Es können vergrößerte Leber und Milz auftreten, des weiteren Herz- und Lungenkomplikationen. MPS VII verursacht oft eine charakteristische Physiognomie und kann progressive Skelettdysplasie auslösen sowie zu Kleinwuchs, Kyphose und Gelenksteifigkeit führen. Weitere mögliche Symptome der MPS VII sind Katarakte, Hornhauteintrübungen, Hernien und nachlassendes Hörvermögen. In schwereren Fällen können auch Entwicklungsverzögerungen auftreten. Bei schwerer Verlaufsform kann die MPS VII sich bereits bei Neugeborenen durch einen Hydrops fetalis (schweres generalisiertes Ödem) äußern. Diese Flüssigkeitsansammlungen enden bei mehr als der Hälfte der Betroffenen bis zum Kleinkindesalter tödlich. Bei einigen Erwachsenen kann MPS VII mit minderschwerem Verlauf auftreten, jedoch führen die schweren zellulären und organischen Dysfunktionen bei MPS VII oft schon im Heranwachsendenalter oder bei jungen Erwachsenen zum Tode.
Weltweit sind ca. 200 Patienten mit MPS VII bekannt.
Diagnose
Neben dem klinischen Bild sind im Labor erhöhte Glykosaminoglykane im Urin nachweisbar. Die Diagnose wird mittels eines Enzymtests gestellt und kann noch durch einen Gentest, der die Mutation im GUSB-Gen nachweist, bestätigt werden.
Symptome
Die Anzeichen der MPS VII können bei einzelnen Betroffenen sehr unterschiedlich ausfallen. Es gibt ein weites Spektrum an möglichen Symptomen. Einige Patienten haben nur leichte Symptome, andere haben schwerere Symptome.
MPS VII betrifft zahlreiche Organsysteme. Die Symptome treten meist schon im jungen Kindesalter in Erscheinung. Folgende Symptome können auftreten:
Komplikationen der Atemwege, Schlafapnoe, häufige Atemwegsinfektionen und Husten
Hornhauteintrübungen und Katarakte
Häufige Ohrinfektionen und Hörverlust
Entwicklungsverzögerungen und geistige Behinderung
Progression der MPS VII
Skelettale Probleme nehmen im Alter meist zu.
MPS VII verläuft progredient. Patienten mit MPS VII erleiden mit zunehmendem Alter häufig Gelenkdeformierungen und schmerzhafte Kontrakturen, die die Beweglichkeit einschränken.
Bei vielen Patienten entwickeln sich:
Abnormale Knochen- und Knorpelstrukturen (Dysostosis multiplex), betroffen sind vor allem Brustkorb, Rücken, Nacken, Knie, Hüften und Hände
Einengung des Wirbelkanals (Spinalstenose) mit möglicher Schädigung des Spinalkanals
Die Lebenserwartung von Patienten mit MPS VII hängt vom Schweregrad der Symptome ab.
Behandlung
In Europa ist eine Enzymersatztherapie (Vestronidase alfa, rhGUS) zugelassen.
Symptombehandlung
Durch unterstützende Maßnahmen kann die Lebensqualität vieler Patienten mit MPS VII verbessert werden.
Das Ziel unterstützender Maßnahmen ist die Milderung der Symptome, um Patienten zu helfen, im Leben besser zurecht zu kommen. Der Arzt sollte unterstützende Maßnahmen koordinieren – dabei wirken oft verschiedene Spezialisten mit:
Neurologen
Orthopäden
Kardiologen
Pneumologen
HNO-Ärzte
Physiotherapeuten
Es wird bei MPS VII empfohlen, sich zu bewegen und einen guten Gesundheitszustand aufrechtzuerhalten, um die Gelenkfunktion und Beweglichkeit lange zu erhalten.
Physikalische Therapie, Physiotherapie und Hydrotherapie kann die Gelenkfunktion aufrechterhalten und die Beweglichkeit verbessern.
In der Behandlung der Symptome können folgende Methoden zum Einsatz kommen. Viele Komplikationen erfordern operative Eingriffe:
Skelettale Manifestationen
Orthopädische Operationen
Komplikationen der Atemwege
Entfernung der Mandeln und/oder Polypen
Verbesserung der nächtlichen Atmung
CPAP (Continuous Positive Pressure Ventilation)
BiPAP (Bi-level Positive Airway Pressure)
Tracheotomie
Asthma-Medikamente
Antibiotika
Schleimhautabschwellende Medikamente
Gelenkversteifungen
Entzündungshemmende Medikamente
Physikalische Therapie, Hydrotherapie
Schienen
Kardiovaskuläre Komplikationen
Medikamentöse Behandlung bei schweren Herzklappenerkrankungen
Operativer Ersatz von Herzklappen
Gastrointestinale Komplikationen
Antibiotika
Ernährungsanpassung
Gastrostomiesonde
Gehörverlust
Hörgerät
Behandlung des Paukenergusses durch Trommelfellschnitt (Myringotomie, reduziert Druck und erleichtert Flüssigkeitsabfluss) und Einsatz eines Paukenröhrchens
Hornhauteintrübung
Hornhauttransplantation
Korrigierende Linsen
Karpaltunnelsyndrom
Operativer Eingriff
Hydrocephalus (Flüssigkeitsansammlung im Gehirn)
Regelmäßige Überwachung
Ventrikuloperitonealer Shunt
Knochenmarktransplantation und Enzymersatztherapie kann zur Behandlung einiger Mukopolysaccharidosen eingesetzt werden.
Knochenmarktransplantation, auch als Hämatopoetische Stammzelltransplantation bezeichnet (Hematopoietic Stem Cell Transplant, HSCT)
HSCT, eine Form der Blutzelltransplantation, ist bei einigen anderen Mukopolysaccharidosen erfolgreich eingesetzt worden. Bei der MPS VII ist HSCT nicht gut etabliert, aber auch schon in einigen Fällen durchgeführt worden. Wenn Patienten mit MPS VII eine HSCT bekommen, erhalten sie Blutzellen von Spendern, die das fehlende Enzym β–Glucuronidase herstellen können. Dadurch können sich die Symptome bessern.
Sprechen Sie mit Ihrem Arzt über die Risiken und den Nutzen einer HSCT.
Enzymersatztherapie (Enzyme replacement therapy, ERT)
Um den Enzymmangel zu beheben, wird bei bestimmten Mukopolysaccharidosen das fehlende Enzym intravenös verabreicht. Bei MPS I, II, IVA, VI und VII kann eine Enzymersatztherapie die Symptome lindern und die Krankheitsprogression verlangsamen.
Überblick
Krankheit
Mukopolysaccharidose Typ VII (MPS VII)
Prävalenz
Laut IQWiG sind in Deutschland 2-7 Patienten mit MPS VII zu erwarten1
Krankheitsmechanismus
Speicherung von Glykosaminoglykanen aufgrund eines Enzymmangels
Symptome
Kleinwuchs, “grobe” Physiognomie, vergrößerte Mandeln, Polypen, vergrößerte Leber und Milz, Hernien, Knochenschmerzen, Gelenksteifigkeit, Karpaltunnelsyndrom, Kardiovaskuläre Komplikationen, Komplikationen der Atemwege, Schlafapnoe, häufige Atemwegsinfektionen und Husten, Hornhauteintrübung und Katarakte, häufige Ohrinfektionen und Hörverlust, Entwicklungsverzögerungen und geistige Behinderung
National MPS Society. A Guide to Understanding MPS VII: Sly Syndrome.
Society for Mucopolysaccharide Diseases. Guide to Understanding Mucopolysaccharidosis VII (MPS VII) Sly.
Lehman TJA, Miller N, Norquist B, et al. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50:v41-v48.
New South Wales Government. Centre for Genetics Education. Autosomal recessive inheritance—traditional patterns of inheritance 1. Fact Sheet 8.www.genetics.edu.au.
Vaux KK. Genetics of Mucopolysaccharidosis Type VII Treatment & Management: Medical Care, Surgical Care, Consultations. Medscape Drugs, Diseases & Procedures. 2011.
Valayannopoulos V, Wijburg FA. Therapy for the mucopolysaccharides. Rheumatology. 2011;50:v49-v59.
Society for Mucopolysaccharide Diseases. Guide to Understanding Mucopolysaccharidosis VII (MPS VII) Sly.
Fox JE, Volpe L, Bullaro J, et al. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol Genet Metab. 2015;114:203-208.
Diese Website verwendet Cookies, um Ihnen einen reaktionsschnelleren und personalisierteren Service zu bieten. Indem Sie unten auf “Akzeptieren” klicken, stimmen Sie der Verwendung von Cookies auf dieser Website zu. Bitte lesen Sie unsere Cookie-Richtlinie and Datenschutzrichtlinie Weitere Informationen zur Verwendung von Cookies auf dieser Website finden Sie hier.